
敬告:CTD已上路,划重点的来了 之二 药学CTD格式到底变了没?(二)
链接:关于发布《M4:人用药物注册申请通用技术文档(CTD)》模块一文件及CTD中文版的通告(2019年第17号)
http://www.nmpa.gov.cn/WS04/CL2138/336252.html
其中附件CTD中文版,就包括了M4(R4)中文版、M4Q(药学/质量)、M4S(非临床/安全性)、M4E(临床/ 有效性)。
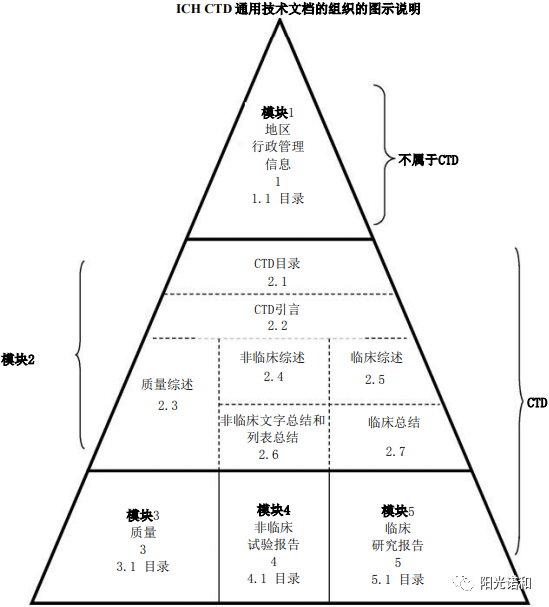
模块二、模块三、模块四、模块五部分CTD格式是一直都在的,只不过是英文版,中文确定版是2019年17号文发布出来的,所以除了模块一需要征求意见外,其他部分都是可以直接落地执行的,并且2018年02月01日起,化学药品注册分类1类、5.1类以及治疗用生物制品1类和预防用生物制品1类申报上市的注册申请已经强制使用《M4:人用药物注册申请通用技术文档(CTD)》,所以对于新药、进口申报申请人,CTD格式并不陌生也更不遥远……
那么药学部分从2010年开始执行所谓的CTD格式到2016年的80号文,再到2019年的M4格式,到底变了没?好嘞,接下来咱们一起比一比、看一看、论一论吧,说在前,我们还是才疏学浅,属于非技术人士,所以不到位、不专业的地方颇多,仅仅是抛砖引玉,希望各位大人们、大咖们海涵海涵……更加欢迎大家来拍砖,多谢多谢!
如果想收藏的亲们,勿急勿急勿急,最后会有个完整版发送的,哈哈
三、 “模块3质量”与80号文药学资料对比:
模块 3:质量
3.1.模块 3 的目录
3.2.主体数据
3.2.S 原料药(名称,生产商)
3.2.S.1 基本信息(名称,生产商)
3.2.S.1.1 药品名称(名称,生产商)
3.2.S.1.2 结构(名称,生产商)
3.2.S.1.3 基本性质(名称,生产商)
3.2.S.2 生产(名称,生产商)
3.2.S.2.1 生产商(名称,生产商)
3.2.S.2.2 生产工艺和工艺控制(名称,生产商)
3.2.S.2.3 物料控制(名称,生产商)
3.2.S.2.4 关键步骤和中间体的控制(名称,生产商)
3.2.S.2.5 工艺验证和/或评价(名称,生产商)
3.2.S.2.6 生产工艺的开发(名称,生产商)
3.2.S.3 特性鉴定(名称、生产商)
3.2.S.3.1 结构和理化性质(名称,生产商)
3.2.S.3.2 杂质(名称,生产商)
3.2.S.4 原料药的质量控制(名称,生产商)
3.2.S.4.1 质量标准(名称,生产商)
3.2.S.4.2 分析方法(名称,生产商)
3.2.S.4.3 分析方法的验证(名称,生产商)
3.2.S.4.4 批分析(名称,生产商)
3.2.S.4.5 质量标准制定依据(名称,生产商)
3.2.S.5 对照品/标准品(名称,生产商) .
3.2.S.6 包装系统(名称,生产商)
3.2.S.7 稳定性(名称,生产商)
3.2.S.7.1 稳定性总结和结论(名称,生产商)
3.2.S.7.2 批准后稳定性研究方案和承诺(名称,生产商)
3.2.S.7.3 稳定性数据(名称,生产商) ..
3.2.P 制剂(名称,剂型)
3.2.P.1 剂型及产品组成(名称,剂型)
3.2.P.2 产品开发(名称,剂型).
3.2.P.2.1 处方组成(名称,剂型)
3.2.P.2.1.1 原料药(名称,剂型)
3.2.P.2.1.2 辅料(名称,剂型)
3.2.P.2.2 制剂(名称,剂型)
3.2.P.2.2.1 处方开发过程(名称,剂型)
3.2.P.2.2.2 过量投料(名称,剂型)
3.2.P.2.2.3 制剂相关特性(名称、剂型)
3.2.P.2.3 生产工艺的开发(名称、剂型)
3.2.P.2.4 包装系统(名称,剂型)
3.2.P.2.5 微生物属性(名称,剂型)
3.2.P.2.6 相容性(名称,剂型)
3.2.P.3 生产(名称,剂型)
3.2.P.3.1 生产商(名称,剂型)
3.2.P.3.2 批处方(名称,剂型)
3.2.P.3.3 生产工艺和工艺控制(名称、剂型)
3.2.P.3.4 关键步骤和中间体的控制(名称、剂型)
3.2.P.3.5 工艺验证和/或评价(名称、剂型)
3.2.P.4 辅料的控制(名称,剂型)
3.2.P.4.1 质量标准(名称,剂型)
3.2.P.4.2 分析方法(名称,剂型)
3.2.P.4.3 分析方法的验证(名称,剂型)
3.2.P.4.4 质量标准制定依据(名称,剂型)
3.2.P.4.5 人源或动物源辅料(名称,剂型)
3.2.P.4.6 新型辅料(名称,剂型)
3.2.P.5 制剂的质量控制(名称,剂型)
3.2.P.5.1 质量标准(名称,剂型)
3.2.P.5.2 分析方法(名称,剂型)
3.2.P.5.3 分析方法的验证(名称,剂型)
3.2.P.5.4 批分析(名称,剂型)
3.2.P.5.5 杂质分析(名称,剂型)
3.2.P.5.6 质量标准制定依据(名称,剂型)
3.2.P.6 对照品/标准品(名称,剂型)
3.2.P.7 包装系统(名称,剂型)
3.2.P.8 稳定性(名称,剂型)
3.2.P.8.1 稳定性总结和结论(名称、剂型)
3.2.P.8.2 批准后稳定性研究方案和承诺(名称、剂型)
3.2.P.8.3 稳定性数据(名称,剂型)
3.2.A 附录
3.2.A.1 设施和设备(名称,生产商)
3.2.A.2 外源因子的安全性评价(名称、剂型、生产商)
3.2.A.3 辅料
3.2.R 区域性信息
3.3 参考文献
内涵引文(写在最前):“模块3质量”中3.1是模块3的目录,3.2是药品质量的主体数据,3.3是模块3的参考文献集合。其中,3.2主体数据中的3.2.S原料药和3.2.P制剂在80号文中有体现,但并不完全相同,80号文针对不同颗粒度提出的具体要求和细节,也都是带有一定的中国特色,跟中国药学研发水平与生产企业当前状况息息相关。
如果写资料时候拿捏不准,对于分到哪个颗粒度或者怎么来书写、讲故事更合适有疑问的话,推荐大家撰写资料或者设计试验前结合着《M4Q(R1)指导原则》看下《M4Q(R1)问答文件》,肯定会受益匪浅茅塞顿开的,开卷有益呦,千万别错过!更多的ICH指导原则详见中文版官方出处,本文下边会详细有每个模块、颗粒度所用的指导原则,均可在官方出处找到,大家一定要更多习惯去应用使用,时时常常去转转看看……
链接:
(1)关于发布《M4:人用药物注册申请通用技术文档(CTD)》模块一文件及CTD中文版的通告(2019年第17号)
http://www.nmpa.gov.cn/WS04/CL2138/336252.html
(2)ICH指导原则中文版官方出处
http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/0/1
(一)3.2.S原料药
对比说明:我们对比了ICH M4Q(R1)指导原则和80号文具体资料要求,发现原料药部分的3.2.S.5对照品/标准品、3.2.S.3特性鉴定没有明显差异。存在差异的内容如下:
1、3.2.S.1基本信息(名称,生产商)
|
M4Q(R1) |
80号文 |
|
3.2.S.1.1 药品名称(名称,生产商) |
3.2.S.1.1药品名称 |
|
3.2.S.1.2 结构(名称,生产商) |
3.2.S.1.2结构 |
|
3.2.S.1.3基本性质(名称,生产商) |
3.2.S.1.3理化性质 |
对比说明:对比资料要求,在M4Q(R1)中,有要求提供“其他相关性质”,故3.2.S.1.3的名称为“基本性质”。
参考ICH指导原则:Q6A和Q6B
2、3.2.S.2生产(名称,生产商)
|
M4Q(R1) |
80号文 |
|
3.2.S.2.1生产商(名称,生产商) |
3.2.S.2.1生产商 |
|
3.2.S.2.2生产工艺和工艺控制(名称,生产商) |
3.2.S.2.2生产工艺和过程控制 |
|
3.2.S.2.3物料控制(名称,生产商) |
3.2.S.2.3物料控制 |
|
3.2.S.2.4关键步骤和中间体的控制(名称,生产商) |
3.2.S.2.4关键步骤和中间体的控制 |
|
3.2.S.2.5工艺验证和/或评价(名称,生产商) |
3.2.S.2.5工艺验证和评价 |
|
3.2.S.2.6生产工艺的开发(名称,生产商) |
3.2.S.2.6生产工艺的开发 |
对比说明:相比较,M4Q(R1)中要求在“3.2.S.2.1生产商”中“应提供每个生产商的名称、地址和职责,包括合同商、生产和检验所涉及的各个拟定生产场所或设施。”这一点是考虑到生产环节可能放在不同生产商,并且同一环节可能存在多个生产商,故此处强调了“每个”及“职责”。
在“3.2.S.2.6生产工艺的开发”中“应说明并论述用于生产非临床批次、临床批次、放大批次、中试规模批次以及生产规模批次(如适用)的原料药生产工艺和/或生产场地发生的主要变更。”此处有强调需要将“非临床批次、临床批次”的工艺也一并进行讨论。
参考ICH指导原则:Q3A、Q5A、Q5B、Q6B和Q11。
3、3.2.S.4原料药的质量控制(名称,生产商)
|
M4Q(R1) |
80号文 |
|
3.2.S.4.1质量标准(名称,生产商) |
3.2.S.4.1质量标准 |
|
3.2.S.4.2分析方法(名称,生产商) |
3.2.S.4.2 分析方法 |
|
3.2.S.4.3分析方法的验证(名称,生产商) |
3.2.S.4.3分析方法的验证 |
|
3.2.S.4.4批分析(名称,生产商) |
3.2.S.4.4 批检验报告 |
|
3.2.S.4.5质量标准制定依据(名称,生产商) |
3.2.S.4.5 质量标准制定依据 |
对比说明:相比较,关于3.2.S.4.4,M4Q(R1)中强调“批分析”,而80号文强调“检验报告”。其他资料要求相似。
参考ICH指导原则:Q2A、Q2B、Q3A、Q3C、Q6A和Q6B
4、3.2.S.6包装系统(名称,生产商)
|
M4Q(R1) |
80号文 |
||||
|
3.2.S.6包装系统(名称,生产商) |
应提供包装系统的说明,包括各初级包装组件结构材料的鉴别及其质量标准。质量标准应包括性状和鉴别(以具体的图例表示关键尺寸,如适用)。如适用,应提供非药典方法(包括相应的验证)。 对于非功能性次级包装组件(如不提供额外保护的包材),仅提供简要说明。对于功能性次级包装组件,应提供更多的信息。 应结合如材料的选择、防潮和避光、包装材料与原料药的相容性,包括容器的吸附和浸出,和/或包装材料的安全性等进行包材适用性的论述。 |
3.2.S.6包装材料和容器 |
(1)包材类型、来源及相关证明文件:提供包材的批准证明文件及检验报告(可来自包材生产商或供应商)。 (2)阐述包材的选择依据。 (3)提供针对所选用包材进行的支持性研究的相关资料。 |
||
对比说明:相比较,资料要求有所不同,M4Q(R1)中更强调包装的整体性,作为一个系统,包括初级包装同时包括非功能性次级包装组件。80号文中未对此进行说明,但要求提供包材证明性文件。
内涵文:国外对合法性文件(证明性文件)是弱化的,不像中国有各种各样的证书、各式各类的文件、大大小小的公章……反反复复都是要证明出来真实存在、来源纯正,合法性在80号文药学研究资料中也是一种独特的存在,在ICH M4格式资料中合法性证明性文件则都放在了行政区域文件即模块1部分,让药学研究资料因为技术而存在,研究深入而纯粹了,探讨更多的是适应性、可行性,而非真实性、合法性了,但原料药的包材还是建议去做登记而后关联审评,更好的保证安全性与可行性。
5、3.2.S.7稳定性(名称,生产商)
|
M4Q(R1) |
80号文 |
|
3.2.S.7.1稳定性总结和结论(名称,生产商) |
3.2.S.7.1稳定性总结 |
|
3.2.S.7.2批准后稳定性研究方案和承诺(名称,生产商) |
3.2.S.7.2上市后稳定性承诺和稳定性方案 |
|
3.2.S.7.3稳定性数据(名称,生产商) |
3.2.S.7.3稳定性数据 |
对比说明:相比较,M4Q(R1)中对3.2.S.7.3有要求“应提供获得稳定性数据所采用的分析方法及其方法学验证信息”。其他资料要求相似。
参考ICH指导原则:Q1A、Q1B、Q2A、Q2B和Q5C
(二)3.2.P制剂
内涵引文(写在最前):
由于M4Q(R1)中的3.2.P.4不再包含原料药,制剂中涉及原料药的内控研究部分或者原料药的深入研究探索部分将视情况而定了。
1) 原料药已经在原辅包登记平台上登记备案公示成功,制剂申请人对原料药也没有再补充研究,那么原料部分可以不提交任何资料;
2) 原料药已经在原辅包登记平台上登记备案公示成功,制剂申请人对原料药有再补充研究,那么针对补充研究的资料,按照3.2.S中编号提供相应资料,不涉及的写不适用;
3) 原料药不强制在原辅包登记平台上,那么原料药的药学研究资料2.3.S和3.2.S需要按照原料要求提供全部资料,放入制剂中一并申报递交。
介于制剂需要对原料药进行全面的评估与审计,我们建议制剂申请人最好请原料药供应商提供2.3.S及3.2.S,至少提供不涉密的公开部分,评估判断是否做深入研究,将多少原料资料放入制剂申报资料中。
对比说明:我们对比了ICH M4Q(R1)指导原则和80号文具体资料要求,发现制剂部分的3.2.P.1剂型及产品组成、3.2.P.5制剂的质量控制、3.2.P.6对照品/标准品没有明显差异。存在差异的内容如下:
1、3.2.P.2产品开发(名称,剂型)
|
M4Q(R1) |
80号文 |
|
3.2.P.2.1处方组成(名称,剂型) 3.2.P.2.1.1原料药(名称,剂型) 3.2.P.2.1.2辅料(名称,剂型) |
3.2.P.2.1处方组成 3.2.P.2.1.1原料药 3.2.P.2.1.2辅料 |
|
3.2.P.2.2制剂(名称,剂型) 3.2.P.2.2.1 处方开发过程(名称,剂型) 3.2.P.2.2.2 过量投料(名称,剂型) 3.2.P.2.2.3 制剂相关特性(名称、剂型) |
3.2.P.2.2 制剂研究 3.2.P.2.2.1处方开发过程 3.2.P.2.2.2制剂相关特性 |
|
3.2.P.2.3 生产工艺的开发(名称、剂型) |
3.2.P.2.3生产工艺的开发 |
|
3.2.P.2.4 包装系统(名称,剂型) |
3.2.P.2.4包装材料/容器 |
|
3.2.P.2.5 微生物属性(名称,剂型) |
—— |
|
3.2.P.2.6 相容性(名称,剂型) |
3.2.P.2.5相容性 |
对比说明:M4Q(R1)中的P.2颗粒度有调整且内容相较80号文也有一定的差异。
对“3.2.P.2.1.1原料药”增加了对于复方制剂的要求,“应论述各原料药之间的相容性”。
相较80号文,M4Q(R1)中将“3.2.P.2.2.2 过量投料(名称,剂型)”从“3.2.P.2.2.1 处方开发过程”中独立出来,作为一个层级单独存在。
“3.2.P.2.4 包装系统”中重点要求应对制剂贮藏、运输和使用时所用的包装系统的适用性进行论述。
内涵文:与3.2.P.7对比,该部分内容是从制剂出发,讨论包材80号文中要求的包材证明性文件未在此处体现,而是ICH-M4(CTD)格式要求将包材证明性文件资料放在M1部分。“微生物属性”也作为单独层级存在于P.2中。
参考ICH指导原则:Q6A和Q6B、Q8、Q9、Q10
2、3.2.P.3生产(名称,剂型)
|
M4Q(R1) |
80号文 |
|
3.2.P.3.1 生产商(名称,剂型) |
3.2.P.3.1生产商 |
|
3.2.P.3.2 批处方(名称,剂型) |
3.2.P.3.2 批处方 |
|
3.2.P.3.3 生产工艺和工艺控制(名称、剂型) |
3.2.P.3.3 生产工艺和工艺控制 |
|
3.2.P.3.4关键步骤和中间体的控制(名称、剂型) |
3.2.P.3.4 关键步骤和中间体的控制 |
|
3.2.P.3.5工艺验证和/或评价(名称、剂型) |
3.2.P.3.5 工艺验证和评价 |
|
—— |
3.2.P.3.6临床试验/BE试验样品的生产情况(80号文中化药4、5.2类的要求) |
对比说明:相比较,与原料药类似,在M4Q(R1)中对“3.2.P.3.1生产商”中“应提供每个生产商的名称、地址和职责,包括合同商、生产和检验所涉及的各个拟定生产场所或设施。”这一点是考虑到生产环节可能放在不同生产商,并且同一环节可能存在多个生产商,故此处强调了“每个”及“职责”。
“3.2.P.3.5工艺验证和/或评价(名称、剂型)”中也未像80号文提及需要工艺验证承诺书,而是要求“应提供验证和/或评价研究的说明、文件和结果(例如灭菌工艺、无菌工艺或灌装的验证)”。
80号文化药4、5.2类的资料要求中,还有“3.2.P.3.6临床试验/BE试验样品的生产情况”该层级,但M4Q(R1)中仅在3.2.P.2.3 生产工艺的开发(名称、剂型)层级中要求“应明确生产关键临床批次所用的生产工艺与3.2.P.3.3所述的生产工艺之间的差异,并论述这些差异是否对制剂性能产生影响”。
参考ICH指导原则:Q2A、Q2B、Q6A和Q6B。
3、3.2.P.4辅料的控制(名称,剂型)
|
M4Q(R1) |
80号文 |
||||
|
3.2.P.4辅料的控制(名称,剂型) |
3.2.P.4.1质量标准(名称,剂型) |
3.2.P.4原辅料的控制 |
提供原辅料的来源、相关证明文件以及执行标准。 如制剂生产商对原料药、辅料制定了内控标准,应分别提供制剂生产商的内控标准以及原料药/辅料生产商的质量标准。 提供原料药、辅料生产商的检验报告以及制剂生产商对所用原料药、辅料的检验报告。 |
||
|
3.2.P.4.2分析方法(名称,剂型) |
|||||
|
3.2.P.4.3 分析方法的验证(名称,剂型) |
|||||
|
3.2.P.4.4质量标准制定依据(名称,剂型) |
|||||
|
3.2.P.4.5人源或动物源辅料(名称,剂型) |
—— |
||||
|
3.2.P.4.6新型辅料(名称,剂型) |
—— |
||||
对比说明:M4Q(R1)与80号文对比,在3.2.P.4层级下M4Q(R1)不包含原料药的内容,只有辅料,且将内容进行了细化,其中3.2.P.4.1~3.2.P.4.4和80号文中的内容有一定的对应关系,但分出了“3.2.P.4.5人源或动物源辅料”、“3.2.P.4.6新型辅料”两部分内容。同时辅料证明性文件资料放在模块1部分。
内涵文:同样一脉相承,证明性合法性真实性的考量点在模块1部分了,给药学一个展示技术才华的平台!
参考ICH指导原则:Q2A、Q2B、Q3C、Q5A、Q5D和Q6B。
4、3.2.P.7包装系统(名称,剂型)
对比说明:该部分为M4Q(R1)特有层级,强调药品包装的整体性,从包装材质和材料角度阐述系统对药的影响。要求提供各初级包装组件、次级包装组件(非功能性或功能性)都进行说明。
内涵文:需要与3.2.P.2.4对比侧重后书写,千万!
3.2.P.8稳定性(名称,剂型)
|
M4Q(R1) |
80号文 |
|
3.2.P.8.1稳定性总结和结论(名称、剂型) |
3.2.P.7.1稳定性总结 |
|
3.2.P.8.2批准后稳定性研究方案和承诺(名称、剂型) |
3.2.P.7.2上市后的稳定性承诺和稳定性方案 |
|
3.2.P.8.3稳定性数据(名称,剂型) |
3.2.P.7.3稳定性数据 |
对比说明:由于增加了3.2.P.7包装系统,稳定性在M4Q(R1)中的编号为3.2.P.8,其中对3.2.P.8.3有要求“应提供获得稳定性数据所采用的分析方法及其方法学验证信息”。其他资料要求相似。
内涵文:不仅仅是表格数据全部堆放,更多需要侧重于探讨总结论述,从而确定贮藏条件、有效期、以及肯定包装系统的可行性。
参考ICH指导原则:Q1A、Q1B、Q2A、Q2B、Q3B、Q5C和Q6A
(三)3.2.A附录和3.2.R区域性信息
对比说明:3.2主体数据中还有3.2.A附录和3.2.R区域性信息。3.2.A附录包含3.2.A.1设施和设备,M4Q(R1)中对该部分的要求是针对生物技术产品的;3.2.A.2是外源因子的安全性评价,在该部分内容中需要与外源因子有关的潜在污染的风险评估信息;3.2.A.3辅料M4Q(R1)中未做要求。
3.2.R区域性信息中针对每个区域的任何其他原料药和/或制剂信息,应在申报资料的R章节提供。这部分内容需要申请人根据本国的区域性指导原则和/或向各监管方咨询情况撰写。根据我们以往的进口品种申报情况来看,CDE有提出过要求在制剂申报进口上市时,需要在R章节提供分析方法验证报告及典型图谱。
内涵文:A与R以后如何被利用,目前未知,既然是可以有各地特色的东西,不妨大胆猜测和预想下未来的A与R会否变成所有研究附件和图谱的堆积处呢?让我们拭目以待吧。
四、 药学与临床:
对比说明:如果是单纯说M4的药学相关部分,应该说已经结束了,但是我们却在模块5中找到了一个特别的存在。在模块5 临床研究报告和相关信息的组织指南中,“5.3.1.3体外-体内相关性研究”项下有要求“提供BA信息的体外溶出度研究报告,包括用于探寻体外数据与体内数据相关性的研究,应放在第5.3.1.3节中”。
|
模块 |
一级标题 |
二级标题 |
三级标题 |
具体要求 |
|
模块5临床研究报告和相关信息的组织指南 |
5.3临床研究报告 |
5.3.1生物药剂学研究报告 |
5.3.1.3体外-体内相关性研究 |
提供BA信息的体外溶出度研究报告,包括用于探寻体外数据与体内数据相关性的研究,应放在第5.3.1.3节中。 用于批次质量控制和/或批次释放的体外溶出度试验报告应放在CTD的质量部分。 |
内涵引文: ——此处纯是个人乱想,大家拍砖!
什么叫体外数据与体内数据相关性的研究呢?仁者见仁智者见智,这个是一个比较新的领域,简称IVIVC,体内体外相关性,IVIVC表示制剂体外特征(例如,药物释放曲线)与其体内性能(例如浓度-时间曲线)之间的数学关系,目的是使用其体外溶出数据准确地预测药物的体内血药浓度-时间曲线。使用经过验证的IVIVC来获得生物等效豁免,即通过使用溶出试验来替代体内生物等效性试验(BE),从而在产品开发和药品整个生命周期中节省了时间和成本。药物的IVIVC评价体系较为完善,以BCS为其理论基础,以溶出曲线和渗透性试验为主要的体外试验方法,以动物或人体为体内试验对象,通过数学模型可以建立体内外数据的相关关系。通过模型药物对数学模型进行验证,可以筛选出与体内试验具有较强相关性的体外试验条件。
这么说起来好高大上,不过换言之理解可能就简单多了,我们需要将溶出曲线的对比及试验结果放于此处,不妨可以参考“2016年第120号通告附件:化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)”中的“11.溶出曲线相似性评价”资料要求进行撰写对比,但重点放在从体外论述体内的可行性。
“2016年第120号通告附件:化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)”中的“11.溶出曲线相似性评价”:
11.溶出曲线相似性评价
参照《普通口服固体制剂溶出度试验技术指导原则》和《普通口服固体制剂溶出曲线测定与比较指导原则》,建立能客观反映制剂特点的溶出试验方法,应具有适当的灵敏度和区分力。
此处感觉像是体外的药学研究工作者,借着体外的相关数据和结果来对临床审评员的一次郑重表白,表白的位置有了,如何表白能够打动人心赢得赞誉,就得看是哪家企业哪个品种打算采用什么形式怎么去做了,仿制药无外乎都是要征求临床审评员同意给减免临床。
想表白想减免BE,欲戴其冠必承其重,我们真的踏踏实实做好多好多,不要等着错过了才追悔莫及,更加要了解想成功就必须有取舍!
小编随机询问了下药学研发人员:如果有一次机会拿着药学资料跟临床审评员沟通的话,您打算怎么表白?走访的结果由我司网红娟做了最新创作图,仅供大家赏析,博众一笑……
1)没留神的发现还有这个机会呀,马上带着我的溶出曲线极速转弯!
2) 老师你不会不给我这么完美的溶曲免be吧?不会吧不会吧不会吧?
3) 我们自认为已经做了全部的药学研究工作,无坚不摧,无懈可击,但是……结果却是……
因为临床审评老师并没有看到我们药学的资料……啥?!
妖艳小喵(药研小苗):老师,请看我真挚饱含热泪的双眸……
回复:药学溶出没放临床中,我怎么去看到呢?晚了!
综上,从ICH M4及M4Q(R1)指导原则的内容来看,该部分内容囊括了化药、生物制品等,所以具体的质量资料要求内容不多,具体研究内容要参见ICH Quality(质量)部分的指导原则,而80号文是针对于化药的申报资料要求,会根据中国的医药研发特点,对各部分内容进行细化并具体要求,大家想对得上审评员的中国心中国胃,建议咱们干脆拿着M4格式研究着相关内容,按照80号更多的细化细节处来补充完善好,查缺补漏!相信慢慢摸索中前进的医药研发人员们和审评员们都在与时俱进,磨合出来众所公认的模板、资料细节以及相关制度,天空飘来五个字儿:那都不是事儿!
强大如我们,中国硬实力,大家一起补上M4的中国心儿!
敬告:CTD已上路,划重点的来了 系列 之三 一直不上心、总是被要求的非临床CTD如何实现? 敬请期待……
题外话:药学申报资料——M4格式与80号文我们也做了自我感觉还比较充分的对比,形成了M4格式与80号文对比表,不过只是抛砖引玉,大家感兴趣的话,欢迎关注我们、来电或微信咨询洽谈等等

干注册我们是认真的!针对不同品种、不同企业、不同个性我们会量体裁衣、定制专业的注册服务,法规文件的详细分析解读,我们恭候您的垂询,共同探讨,为咱们共同的项目目标,寻求更好的注册解决策略!与时俱进的阳光诺和注册部向您致敬!
研发驱动,创新驱动,平台驱动——阳光诺和是专注于仿制和创新药物研究,致力于以技术服务为核心,推动中国医药行业发展,为客户提供「临床前+临床」全过程药物研发服务的综合型CRO公司。

